






Research article
H.A. Madani,1 R.A. Afify,1 A.A. Abd El-Aal,1 N. Salama 2 and N. Ramy2
دور طفرات الجين HFE في ظهور فرط حمل الحديد لدى حَمَلَة البيتا ثلاسيمية في مصر
حنان علي مدني، ريهام عبد العليم عفيفي، أسماء أحمد عبد العال، نيفين سلامة، نيرمين رامي
الخلاصـة: تهدف دراسة الحالات والشواهد هذه إلى تحديد انتشار الطفرات C282Y و H63D و S65C للجين HFE بين حَمَلَة البيتا ثلاسيمية وتقصي تأثيرها على امتصاص الحديد. وقد أجرت الباحثات تحرِّيات شملت 41 شخصاً حاملاً للبيتا سلاثيمية و40 شخصاً شاهداً من غير المصابين بأي اعتلال هيموغلوبيني، بحثاً عن وجود الطفرات C282Y، H63D، S65C وذلك بإجراء التفاعل السلسلي للبوليميراز، وتعدد أشكال أطوال الشُّدف الـمُقْتَطَعَة. كذلك دُرِست حالة الحديد في هؤلاء الأشخاص وارتباطها بطفرات الجين HFE. وقد كان تواتر أليل الطفرة H63D: %30.5 وتواتر أليل الطفرة S65C: %13.4 وتواتر أليل الطفرة C282Y: %7.3 بين حَمَلَة البيتا ثلاسيمية، و%10.0 و%2.5 و%0 على التوالي في مجموعة الشواهد. وقد وجدت الباحثات تغايُرَ الزيجوت المركّب في عشرة أشخاص من حَمَلَة البيتا ثلاسيمية (%24.4). كما كان مستوى تشبُّع الترانسفيرين مرتفعاً في حالات تغاير الزيجوت المركّب. وقد دلت الدراسة على أن طفرات الجين HFE شائعة الحدوث في مصر بين حَمَلَة البيتا ثلاسيمية مقارنة بالأشخاص الطبيعيين من الشواهد.
ABSTRACT: A case–control study aimed to determine the prevalence of C282Y, H63D and S65C mutations of the HFE gene in β-thalassaemia carriers and investigate their influence on iron absorption. A total of 41 β-thalassaemia carriers and 40 control subjects without haemoglobinopathies were screened for the C282Y, H63D and S65C mutations by polymerase chain reaction-restriction fragment-length polymorphism. The iron status in these subjects was studied and correlated with the HFE gene mutations. H63D, S65C and C282Y allele frequencies were 30.5%, 13.4% and 7.3% respectively in β-thalassaemia carriers and 10.0%, 2.5% and 0.0% respectively in the control group. Compound heterozygosis was found in 10 carriers (24.4%). The transferrin saturation level was high in compound heterozygote cases. Our study has shown that the HFE gene mutations are common in Egypt among β-thalassaemia carriers compared with normal controls.
Rôle des mutations du gène HFE dans l'apparition d'une surcharge en fer chez les porteurs d'une β-thalassémie en Égypte
RÉSUMÉ: La présente étude cas-témoins visait à déterminer la prévalence des mutations C282Y, H63D et S65C du gène HFE chez les porteurs d'une β-thalassémie et à rechercher leur influence sur l'absorption du fer. Au total, 41 porteurs d'une β-thalassémie et 40 sujets témoins ne présentant aucune hémoglobinopathie ont participé à cette étude visant à examiner les mutations C282Y, H63D et S65C du gène HFE par la méthode du polymorphisme de longueur des fragments de restriction d'ADN amplifié. Le bilan martial des participants a été étudié et corrélé aux mutations du gène HFE. La fréquence des allèles H63D, S65C et C282Y était de 30,5 %, 13,4 % et 7,3 % respectivement, chez les porteurs d'une β-thalassémie et de 10,0 %, 2,5 % et 0,0 % respectivement, dans le groupe témoin. Une hétérozygotie composée a été retrouvée chez dix porteurs (24,4 %). Le taux de saturation de la transferrine était élevé chez les cas hétérozygotes composés. Notre étude a démontré que les mutations du gène HFE sont fréquentes en Égypte chez les porteurs d'une β-thalassémie par rapport aux sujets témoins.
EMHJ, 2011, 17(6): 546-551
1Department of Chemical and Clinical Pathology;
2Department of Paediatric Medicine, Faculty of Medicine, University of Cairo, Cairo, Egypt (Correspondence to H.A. Madani:
Introduction
Thalassaemia syndromes are the most commonly inherited single-gene disorders worldwide [1]. About 3% of the world population (150 million) carry the β-thalassaemia genes [2]. In Egypt β-thalassaemia is the most common genetically determined, chronic, haemolytic anaemia with an estimated carrier rate of 9%–10.5% [3]. The β-thalassaemia trait is associated with extremely mild anaemia and red-cell morphological changes; however it may be completely silent with no anaemia or haematological abnormalities [3,4]. It is characterized by mild, ineffective erythropoiesis that can induce excessive iron absorption from the diet because of higher iron requirement for haemoglobin synthesis [5].
β-thalassaemia trait alone does not lead to iron overload, and some gene modifiers and acquired causes are reported to modulate the expression of hereditary haemochromatosis [6]. Haemochromatosis is a common autosomal recessive disorder of iron metabolism, with a prevalence of 1 in 300–500 individuals [7], which can lead to total-body iron overload with secondary tissue damage in a wide range of organs. When β-thalassaemia trait is inherited together with a mutation in the HFE gene, which is associated with hereditary haemochromatosis, iron overload may ensue [8]. The HFE gene is located on the short arm of chromosome 6 at position 21.3; its mutation is a major cause of haemochromatosis. Three missense mutations in the HFE gene are associated with haemochromatosis. About 85% of cases carry cysteine-to-tyrosine substitution at amino acid position 282 in the HFE gene (C282Y) [9]. This mutation is responsible for about 60% of haemochromatosis cases in Mediterranean populations [10], about 15%–20% carry aspartic acid-to-histidine substitution at amino acid position 63 (H63D) [11,12] and a third mutation results from serine-to-cysteine conversion in amino acid position 65 (S65C) [13]. The last 2 mutations are associated with a milder form of haemochromatosis, but the compound heterozygous state of one of them with the C282Y mutation increases the risk of developing iron overload [14,15]. The interaction of HFE mutations with the thalassaemias may have a synergistic effect, increasing the iron intake and storage [9].
The high frequency of β-thalassaemia trait in Egypt led us to design a case–control study to determine the prevalence of C282Y, H63D and S65C mutations of HFE gene in β-thalassaemia carriers and investigate their influence on iron absorption, comparing them with individuals without thalassaemias.
Methods
Sample
This study was carried out during the period between March and December 2007. We studied 2 groups: 41 cases of β-thalassaemia carriers (10 males and 31 females) with a mean (standard deviation) age of 38 [standard deviation (SD) 3.2] years, who were parents of thalassaemia major patients attending the haematology clinic at the new Cairo University children’s hospital; and 40 subjects (15 males and 25 females) with a mean age of 37.2 (SD 2.7) years without haemoglobinopathies, as a control group. Diagnosis of β-thalassaemia trait was defined by low mean corpuscular volume (MCV < 80 fL), low mean corpuscular haemoglobin (MCH 3.5%). Control subjects were characterized by normal haematological parameters.
The institutional ethics committee approved the study and informed consent was obtained from all participants. The exclusion criteria were: subjects with impaired liver function (which could interfere with iron metabolism); blood donors (as their iron status may be decreased as a result of repeated blood donation); and children (because their iron deposition tends to increase with age and may reach moderately high levels in adults).
Laboratory workup
Haematological and iron parameters
Blood samples were collected from all participants and tested at the Chemical and clinical pathology laboratory at the new Cairo University children’s hospital. A 2 mL of blood sample was collected into EDTA tubes for complete blood count using an electronic Coulter counter (Sysmex KX-21N) and haemoglobin electrophoresis on cellulose acetate paper at pH 8.4. A 3 mL blood sample was taken into plain tubes for determination of serum iron and total iron binding capacity (TIBC). Both iron and TIBC were assessed using standard colorimetric kits by automated analyser (Beckman Coulter Synchron CX9 Pro). Normal range for iron was 70–200 µg/dL and for TIBC was 250–435 µg/dL. The transferrin saturation index was calculated using the formula: serum iron/TIBC × 100 (normal range 20%–45%) [16].
Genomic DNA analysis
A 3mL blood sample was collected into EDTA vacutainers for genomic DNA analysis by polymerase chain reaction–restriction fragment-length polymorphism (PCR–RFLP). DNA was extracted from peripheral blood leukocytes by the salting- out procedure [17].
Two primer sets were used for DNA amplification. The first, 5ʹACATGGTTAAGGCCTGTTGC3ʹ and 5ʹGCCACATCTGGCTTGAAATT3ʹ, generates a 207-bp fragment that comprises the H63D and S65C mutation sites. The second, ʹGGGTATTTCCTTCCTCCAACC3ʹ and ʹCTCAGGCACTCCTCTCAACC3ʹ, generates a 441-bp fragment for C282Y analysis. The PCR cycles were conducted in a thermal cycler (Biometra UNO-thermoblock) and consisted of 5 min of initial denaturation at 94 °C, 30 cycles of just 1 min. denaturation at 94 °C , 1 min. annealing at 58 °C and 1 min. extension at 72 °C, followed by 10 min. for final extension at 72 °C. The amplified PCR products were digested with the restriction enzymes BclI, HinfI and RsaI for H63D, S65C and C282Y mutations respectively.
The C282Y mutation creates a new RsaI site. The digested PCR product is cut into 2 fragments of 296 and 145 bp in the normal allele, while in the mutated DNA, 3 fragments of 296, 116 and 29 bp are generated after digestion (Figure 1). The H63D mutation abolishes the BclI recognition site in the 207 bp PCR product; while normal DNA is cut into 2 fragments of 138 and 69 bp the mutated DNA is not cut (Figure 2). The S65C mutation abolishes the HinfI recognition site in the 207 bp PCR product; while normal DNA is cut into 2 fragments of 147 and 60 bp, the mutated DNA is not cut (Figure 3) [7].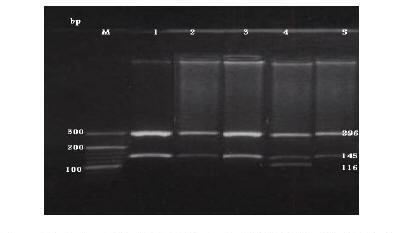
Figure 1 Detection of the HFE C282Y mutation by PCR-RFLP with C282Y primers and digested with restriction enzyme Rsa1. Lane M: DNA marker. Lanes 1, 2, 3 & 5: normal C282Y allele (296 &145 bp). Lane 4: heterozygote alleles (296, 145, 116 & 29 bp).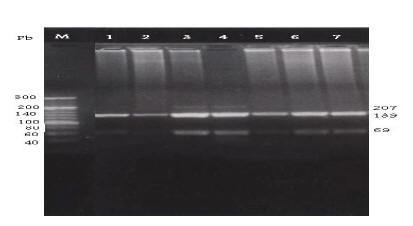
Figure 2 Detection of the HFE H63D mutation by PCR-RFLP with H63D primers and digested with restriction enzyme Bcl1. Lane M: DNA marker. Lanes 1, 5, 6 & 7: normal H63D wild allele digested bands (138 & 69 pb). Lanes 3 & 4: heterozygotes for H63D mutant allele showing normal digested bands (138 & 69 pb) as well as the undigested amplified fragment (207 pb). Lane 2: homozygote for H63D mutant allele show undigested band (207 pb).

Figure 3 Detection of the HFE S65C mutation by PCR-RFLP with S65C primers and digested with restriction enzyme Hinf1. Lane M: DNA marker. Lanes 1, 2, 3 & 6: normal S65C wild allele digested bands (147 & 60 pb). Lanes 4 & 5: heterozygotes for S65C mutant allele showing normal digested bands (147 & 60 pb) as well as the undigested amplified fragment (207). Lane 7: homozygote for S65C mutant allele show undigested band (207 pb).
Allele frequency was calculated using the gene counting method (each individual is represented by 2 alleles; allele frequency is number of mutated alleles/total number of alleles). Chi-squared and Fisher exact tests were used to compare allele and genotype frequency between the β-thalassaemia carriers and the control group. P-valueSPSS, version 11.5.
Results
HFE allelic frequencies and genotypes
The allelic frequencies obtained for HFE gene mutations C282Y, H63D and S65C in the studied groups are presented in Table 1. There were significantly high allelic frequencies of the H63D, S65C and C282Y mutations among β-thalassaemia carriers (30.5%, 13.4% and 7.3% respectively) compared with the controls (10.0%, 2.5% and 0.0%) respectively (Fisher test, P < 0.05).
Table 1 Allele frequencies of H63D, S65C and C282Y mutations among total alleles of beta-thalassaemia carriers and controls without haemoglobinopathies
Mutational analysis for the HFE gene (Table 2) showed that compound heterozygosis was found in 10 of the β-thalassaemia carriers (24.4%): 6 cases were compound heterozygous for the 3 mutations (5 cases heterozygous for the 3 mutations and 1 case heterozygous for H63D/C282Y and homozygous for S65C mutation) and 4 cases were H63D/S65C, while in the control group 2 individuals (5.0%) were H63D/S65C. In β-thalassaemia carriers the genotype wild-type/wild-type (–/–) was the most frequent, accounting for 17 individuals (41.5%).
Table 2 HFE genotype frequencies of H63D, S65C and C282Y mutations among beta-thalassaemia carriers and controls
H63D, S65C and C282Y heterozygote genotype frequencies among β-thalassaemia carriers were 23 (56.1%), 9 (22.0%) and 6 (14.6%) cases respectively while in controls these were 8 (20.0%), 2 (5.0%) and 0 (0.0%) respectively (Table 3).
Table 3 HFE genotype frequencies by sex among beta-thalassaemia carriers and controls
The genotype frequency by sex is also represented in Table 3. The male to female sex ratio in cases of heterozygote C282Y mutation was 6:0, in H63D heterozygote cases was 7:16 and in heterozygote S65C was 6:3.
HFE mutations and iron metabolism
We found 8 patients with high transferrin saturation (> 45%) (7 males and 1 female). Of these, 6 were heterozygous for H63D, S65C and C282Y, 1 patient was heterozygous for H63D and S65C and 1 patient was homozygous for H63D.
We studied the relationship between HFE gene mutations and iron status in β-thalassaemia carriers and found significantly higher allelic frequencies of the H63D, S65C and C282Y mutations among β-thalassaemia carriers (30.5%, 13.4% and 7.3% respectively) compared the controls (10.0%, 2.5% and 0.0% respectively). H63D, S65C and C282Y heterozygote genotype frequencies among β-thalassaemia carriers were 56.1%, 22.0% and 14.6% respectively while in the controls they were 20.0%, 5.0% and 0.0% respectively.
The results of HFE allele frequencies in our study are similar to Panigrahi et al. [18], who reported allelic frequencies of the H63D mutation of 22.5% among patients and 8.1% among controls. The frequency of the C282Y mutation was 0% among patients and controls [18]. Our results also agreed with a screening study for haemochromatosis mutations in β-thalassaemia minor patients from the Islamic Republic of Iran, which indicated significant differences in the frequencies of C282Y and H63D mutants in relation to control individuals, where the H63D and C282Y allele frequencies were 12.9% and 1.6% in patients compared with 8.7% and 0% in controls respectively [9]. The results of our study also agreed with Oliveira et al., who reported allelic frequencies of the H63D, S65C and C282Y mutations among β-thalassaemia carriers of 13.7%, 0.6% and 2.4% respectively compared with 9.5%, 0.9% and 0.3% respectively among controls; the heterozygote genotype frequencies among their patients and controls were not significantly different [7].
In the present study in Egypt, the genotype frequencies and allele frequencies among the control group for H63D, S65C and C282Y are in agreement with other studies in Africa showing an absence of the C282Y mutation in Algeria, Ethiopia and Senegal. The H63D mutation, though absent in Senegalese, was found in about 9% of the chromosomes genotyped among the central Ethiopians and Algerians [19]. Also, our genotype frequencies agreed with the results of a study done in Egypt by Settin et al. who reported H63D and C282Y genotype frequencies as 21.2% and 0.0% respectively in their control subjects [20].
In our study, compound heterozygosis was found in 10 β-thalassaemia carriers: 6 cases were H63D/S65C/C282Y and 4 cases were H63D/S65C, while in the control group 2 individuals were H63D/S65C. Compound heterozygosis for C282Y and H63D seems to predispose to disease expression [21]. The clinical significance of the other forms of compound heterozygosis, such as C282Y and S65C or H63D and S65C, is still controversial [7]. In the present study, the high incidence of the C282Y mutation observed in β-thalassaemia carriers in relation to the control group leads to a concern about the levels of iron deposition in the organs of these patients. Both diseases (haemochromatosis and thalassaemia)affect iron metabolism and the meaning of coinheritance of the 2 mutations is not well understood.
In the present study, high transferring saturation was observed in 8 β-thalassaemia carriers (7 males and 1 female), all of whom carried the 3 mutations. They might be at risk of developing clinical haemochromatosis and strict follow-up would be required to detect early cases.
An interesting point was the male to female sex ratio; where all the heterozygote cases of C282Y mutation were males, a male:female ratio of 6:0, while in the H63D heterozygote cases, the male/female ratio was 7:16 and the heterozygote S65C male/female ratio was 6:3. Male individuals carrying H63D and S65C mutations or one of them in combination with C282Y would be more at risk of developing haemochromatosis than females. Indeed, due to menstruation and pregnancy, females seem to be protected from developing iron overload [22], which can explain why male individuals carrying the mutations have higher transferrin saturation than females. Thus, the risk of developing haemochromatosis is associated with the HFE genotype and the subject’s sex. However, a larger scale population study is needed to confirm this finding as the sample size in this study was small.
Our results confirm the hypothesis that in some way the HFE gene mutations could be implicated in increasing iron storage when interacting with other genetic determinants of β-thalassaemia [23].
Our study has shown that the HFE gene mutations are common among β-thalassaemia carriers in Egypt compared with normal controls. This emphasizes the value of routine screening of the HFE mutations in β-thalassaemia carriers to avoid developing iron overload and to detect early cases. Mass screening for HFE gene mutations among thalassaemia patients is strongly recommended.
References
- Karimi M et al. Prevalence of beta-thalassemia trait and glucose-6-phosphate dehydrogenase deficiency in Iranian Jews. Archives of Medical Research, 2008, 39:212–214.
- Saxena A, Phadke SR. Feasibility of thalassemia control by extended family screening in Indian context. Journal of health, population & nutrition, 2002, 20(1):31–35.
- El Beshlawy A et al. Thalassemia prevalence and status in Egypt. Pediatric Research, 1999, 46:102.
- Krishnamurti L et al.; LaksShaman K et al. Coinheritance of alpha-thalassemia-1 and hemoglobin E/beta zero-thalassemia: practical implications for neonatal screening and genetic counseling. Journal of Pediatrics, 1998, 132:863–865.
- Thein SL. Genetic modifiers of β-thalassaemia . Heamatologica, 2005, 90:649–660.
- Weatherall DJ. Science, medicine, and the future. Single gene disorders or complex traits: lessons from the thalassaemias and other monogenic diseases. BMJ (Clinical Research Ed.), 2000, 321:1117–1120.
- Oliveira TM et al. HFE gene mutations in Brazilian thalassemic patients. Brazilian Journal of Medical and Biological Research, 2006, 39:1575–1580.
- Cançado RD et al. Analysis of HFE gene mutations and HLA-A alleles in Brazilian patients with iron overload. Sao Paulo Medical Journal, 2006, 124:55–60.
- Jazayeri M et al. Frequency of HFE gene mutations in Iranian beta-thalassaemia minor patients. European Journal of Haematology, 2003, 71:408–411.
- Brissot P et al. Clinical aspects of hemochromatosis. Transfusion Science, 2000, 23:193–200.
- Candore G et al. Frequency of the HFE gene mutations in five Italian populations. Blood Cells, Molecules & Diseases, 2002, 29:267–273.
- Bomford A. Genetics of haemochromatosis. Lancet, 2002, 360:1673–1681.
- Mura C, Raguenes O, Férec C. HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis. Blood, 1999, 93:2502–2505.
- Ǻsberg A et al. Hereditary hemochromatosis: the clinical significance of the S65C mutation. Genetic Testing, 2002, 6:59–62.
- Agostinho MF et al. Mutation analysis of the HFE gene in Brazilian populations. Blood Cells, Molecules & Diseases, 1999, 25:324–327.
- Wick M, Pinggera W, Lehmann P. Iron metabolism, anemias, diagnosis and therapy, 4th ed. New York, Springer, Berlin Heidelberg, 2000.
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research, 1988, 16:1215.
- Panigrahi I et al. Evidence for non-HFE linked hemochromatosis in Asian Indians. Indian Journal of Medical Sciences, 2006, 60:491–495.
- Roth M et al. Absence of the hemochromatosis gene Cys282Tyr mutation in three ethnic groups from Algeria (Mzab), Ethiopia, and Senegal. Immunogenetics, 1997, 46:222–225.
- Settin A et al. C282Y and H63D hemochromatosis alleles in Egyptian patients with cirrhosis. Arab Journal of Gastroenterology, 2006, 7:59–63.
- Pietrangelo A. Hereditary hemochromatosis–a new look at an old disease. New England Journal of Medicine, 2004, 350:2383–2397.
- Moirand R et al. Clinical features of genetic hemochromatosis in women compared with men. Annals of Internal Medicine, 1997, 127:105–110.
- Cicilano M et al. Recurrent mutations in the iron regulatory element of L-ferritin in hereditary hyperferritinemia-cataract syndrome. Hematologica, 1999